







Over the past 35 years, treatment of coronary artery disease and acute coronary syndrome has changed drastically. Balloon angioplasty offered an early mechanical solution but carried a high risk for acute complications and subsequent restenosis, mainly from recoil. The bare metal stent (BMS) improved restenosis because of recoil, but still carried a relatively high rate of restenosis because of excessive neointimal hyperplasia. The drug eluting stent (DES) further decreased restenosis by eluting anti-proliferative drugs over time and reducing neointima. However, the first generation of DES raised concerns of increased late and very late stent thrombosis, which were mitigated by prolonged duration of antiplatelet drugs. Newer generations of DES now have lower stent thrombosis risk (<1 % at 1 year),1 particularly when combined with dual antiplatelet therapy (DAPT), but the permanent metal structure in the coronary artery continues to create an ongoing risk of restenosis and stent thrombosis. Late events are related to persistent inflammation, neoatherosclerosis within the stented segment, and inability to restore normal vessel architecture and physiologic function in the presence of a metal structure.
Recently, several new stent constructs have been studied and approved in the United States (US). The SYNERGY™ stent (Boston Scientific)2 is an everolimus-eluting stent (the same drug and platinum chromium alloy as the Promus Premier™ second-generation DES) with an abluminal polymer that absorbs in a few months, leaving behind a BMS. This device has demonstrated non-inferiority to a durable polymer everolimus-eluting stent and is being evaluated for the safety of reducing the duration of antiplatelet therapy to 3 months. Bioresorbable vascular scaffolds (BVS) also use antirestenotic drugs identical to the second-generation DES, but the entire scaffold resorbs over several years. Absorb™ (Abbott) and DESolve™ (Elixir Medical Corporation) are poly-L-lactic acid based BVS that degrade by hydrolysis. DREAMS (Biotronik) is a magnesium-based BVS that degrades by oxidation–reduction reactions. The early experiences and large registry data from Europe led to the Conformité Européene (CE) Mark of Absorb in 2011 and DESolve in 2013. Subsequently, randomized trials in the US, Japan, and China led to the approval of Absorb in these countries. This review will focus on available data for the Absorb BVS and present a rational strategy for adoption of its use for clinical practice in the US.
The goals for a BVS are to provide adequate radial support for a period long enough to avoid early recoil, release anti-proliferative drug for prevention of restenosis, and then resorb early enough to reap the clinical benefits of not leaving a metal device within the artery. Unlike most recent advances in coronary stent technology, the design of the BVS requires the use of thick struts to meet the goal of adequate radial support from a bioresorbable material. For example, the Absorb BVS strut thickness is 150 μm compared with strut thickness of 80–90 μm for contemporary metallic DES. The potential benefits of strut thickness <100 μm of metallic stents for reduced thrombogenicity and neointimal hyperplasia have been demonstrated3 and the potentially negative impact on early and late outcomes of the thicker struts of BVS require consideration. The engineering of the current Absorb BVS allows for an early phase post-percutaneous coronary intervention (PCI; 0–6 months), during which the Absorb BVS provides enough radial strength for mechanical support to maintain vessel patency, while antirestenotic drugs are eluted to prevent restenosis. By 6–12 months, the BVS begins to lose radial strength and reduce mechanical restraints on the vessel to allow healing of the native vessel and restoration of vasoregulation. Once completely resorbed by 3 years, late adaptive remodeling will accommodate new intramural plaque. The restoration of vasomotion and pulsatility has been demonstrated by response to acetylcholine and nitrates.4,5 Optical coherence tomography (OCT) performed 5 years postBVS implantation demonstrates normal intima, media, and adventitia of the coronary artery, the so-called ‘golden tube.’6,7 Other potential practical benefits after the BVS is fully resorbed include the ability to perform coronary artery bypass, ‘unjailing’ of side branch vessels, better coronary computerized tomography imaging without artifacts from metallic stents, and treating restenosis without multiple layers of stents.
Historical Background and Early Data
The first successful use of a fully degradable stent in human was the poly-L-lactic acid based non-drug-eluting Igaki-Tamai scaffold (Kyoto Medical Planning Co., Ltd.).8 Tamai et al. reported initial results from 15 patients in whom 25 scaffolds were successfully implanted in 2000. Long-term data with >10 years of follow-up for 50 patients treated with 84 Igaki-Tamai biodegradable stents showed cumulative target lesion revascularization (TLR) of 16 % after 1 year, 18 % after 5 years, and 28 % after 10 years.9 Despite these promising results, development of the Igaki-Tamai biodegradable stent was discontinued because of the need for a large 8-French guide catheter for delivery and potential vessel wall injury from this heat-treated, self-expandable device.
Refinement of the resorbable scaffold led to development of the Absorb stent. Orniston et al. reported on an early version of the device noting a loss of scaffold area at 6 months likely because of early recoil.10 A subsequent design incorporated changes in manufacturing of the polymer backbone for slower resorption and increased early radial strength. This second-generation BVS was tested in 101 patients with simple atherosclerotic lesions in the ABSORB cohort B trial. Six months’ follow-up of 45 of the 101 patients included in the trial (cohort B1) demonstrated a modest reduction in lumen area as documented by intravascular ultrasound (IVUS) and OCT.11 These favorable results persisted at 12 months’ follow-up of the remaining 56 patients (cohort B2).12 IVUS and OCT examination at baseline and at 12 months’ follow-up showed that the scaffold area remained unchanged. The angiographic late lumen loss was 0.27 ± 0.32 mm with only a 1.94 % relative decrease in minimal lumen area (p=0.12) and without significant changes in mean lumen area as documented by IVUS. OCT examination at 12 months’ follow-up showed that 96.69 % of the struts were covered. Five-year OCT follow-up of 53 patients in ABSORB Cohort B without target lesion revascularization showed no visible struts.6 Major adverse cardiovascular events (MACE) occurred in 11 % with no stent thrombosis. ABSORB Cohort B angiographic and clinical results led to the CE mark of the Absorb BVS in 2011. After CE mark approval, the Gauging Coronary Healing With Bioresorbable Scaffolding Platforms In Europe (GHOST-EU) registry was one of the largest early multicenter evaluations in routine practice, consisting of 10 centers and 1,189 patients.13 All patients with coronary disease suitable for stenting were eligible for the registry. Target lesion failure (TLF, defined as cardiac death, target vessel relatedmyocardial infarction [TV-MI], and target lesion revascularization [TLR]) was 4.4 % at 6 months with 1 % cardiac death and 2.5 % TLR. Diabetes was the only independent predictor of TLF. Paradoxically, TLF was lower (3.2 %) in the first 50 cases at each institution as compared with patients after 50 cases (5.2 %). The investigators hypothesized that this was likely because of more use of intravascular imaging and post-dilation in the earlier cases. Scaffold thrombosis (ScT) rates in the registry were higher than contemporary second-generation DES, with 1.5 % at 30 days and 2.1 % at 6 months. This may have been partly attributable to complex lesions with 27 % bifurcation lesions and 17 % requiring overlapping scaffolds. Alternatively, this may have been related to implantation techniques with only 49 % overall post-dilation.
Randomized Trials
Five trials in the ABSORB series were randomized with Absorb BVS versus Xience (Abbott; metallic second-generation DES), both of which elute everolimus (see Table 1). All trials except TROFI II enrolled mostly stable coronary artery disease patients with some unstable angina patients, and excluded ST-elevation myocardial infarction (STEMI) and non-STEMI patients. ABSORB II immediately followed ABSORB Cohort B, and was powered to evaluate vasomotor function and late lumen loss at 3 years. ABSORB III was the largest randomized trial, designed for US approval and was powered for clinical endpoints. ABSORB TROFI II uniquely included only STEMI patients.
ABSORB II randomized 501 patients. Initial post-PCI imaging showed lower acute lumen gain in the Absorb group based on quantitative coronary angiography (1.15 mm versus 1.46 mm, p<0.0001), and IVUS (2.85mm2 versus 3.6mm2, p<0.0001). MACE rates in the two groups were similar (5 % versus 3 % at 1 year and 7.6 % versus 4.3 % at 2 years).14,15 Likewise, TLF did not significantly differ between the two groups (7.0 % versus 3.0 %, p=0.07 at 2 years). In a substudy, more complex lesions had higher target vessel revascularization (TVR).16 Three-year results of ABSORB II were disappointing for the primary endpoints of vasomotion and late lumen loss.14 Vasomotion assessed by change in mean lumen diameter after intracoronary nitroglycerin was, unexpectedly, not superior in the Absorb group (0.047 mm versus 0.056 mm, psuperiority=0.49), and late lumen loss was actually larger in the Absorb group (0.37mm versus 0.25mm, pnoninferiority=0.78). Although not powered for clinical endpoints, some safety endpoints were concerning. TV-MI (7 % versus 1 %, p=0.01) and definite or probable device thrombosis (3 % versus 0 %, p=0.03) were higher in the Absorb group. In the Absorb group, there were two definite ScT within 1 year and six very late definite ScT (2 %) between 1 and 3 years. All very late ScT were in patients not taking DAPT, raising the question of the appropriate length of DAPT in these patients. Overall device-oriented events were 10 % in Absorb and 5 % in Xience (p=0.04), including TLR in 6 % versus 2 % (p=0.04). 14
ABSORB CHINA was a randomized trial of 480 patients for approval in China based on non-inferiority of angiographic late lumen loss for Absorb BVS compared with Xience. The primary endpoint of in-segment late loss at 1 year was non-inferior (0.19 ± 0.38 mm versus 0.13 ± 0.38 mm; Pnoninferiority=0.01), and TLF at 1 year was similar (3.4 % versus 4.2 %) Definite or probable device thrombosis was also not different (0.4 % versus 0.0 %).17
ABSORB JAPAN was a randomized trial of 400 patients to support approval in Japan based on clinical non-inferiority. The primary endpoint was TLF.18 At 1 year, TLF was similar for Absorb and Xience (4.2 % versus 3.8 %; Pnoninferiority< 0.0001, based on a prespecified 8.6 % non-inferiority margin). Definite or probable device thrombosis was slightly higher than other contemporary DES studies at 1.5 % in both groups. Intracoronary imaging was utilized in approximately 70 % of patients in each group and post-dilation was performed in approximately 80 % in each group. As with ABSORB II and ABSORB CHINA, ABSORB JAPAN showed higher in-device diameter stenosis and lower in-device acute gain in the Absorb group immediately post procedure.
ABSORB III was the pivotal randomized trial with 2,008 patients that led to Food and Drug Administration (FDA) approval of Absorb in the US.19,20 The primary endpoint of TLF in the intention-to-treat analysis was 7.8 % in the Absorb group and 6.1 % in the DES group (pnoninferiority=0.007 based on a prespecified 4.5 % non-inferiority margin). For the per-treatment analysis, the TLF was 7.8 % in the Absorb group versus 5.7 % in the Xience group (Pnoninferiority=0.0183). The three powered secondary outcomes did not significantly differ between the two groups: 1-year angina (18.3 % versus 18.4 %), 1-year all revascularization (9.1 % versus 8.1 %), and 1-year ischemia-driven TVR (5.0 % versus 3.7 %). Several additional secondary outcomes at 1 year were numerically higher in the Absorb group, including all-cause mortality (1.1 % versus 0.4 %), cardiac death (0.6 % versus 0.1 %), all MI (6.9 % versus 5.6 %), and definite or probable device thrombosis (1.5 % versus 0.7 %). The doubling of device thrombosis was concerning. In post-hoc analyses, it was noted that the higher incidence of adverse events in Absorb occurred in smaller vessels. When lesions were separated by reference vessel diameter (RVD) into <2.25 mm (18.8 % of total lesions treated) versus ≥2.25 mm by quantitative coronary angiography (QCA), 1-year events were higher for <2.25 mm vessels treated with Absorb compared to those treated with Xience: TLF 12.9 % versus 8.3 %, device thrombosis 4.6 % versus 1.5 %, and TV-MI 10.0 % versus 4.5 %. In vessels ≥2.25 mm, these endpoints were similar for Absorb and Xience. TLF was 6.7 % versus 5.5 %, device thrombosis 0.9 % versus 0.6 %, and TV-MI 5.2 % versus 4.6 %.19
ABSORB TROFI II randomized 191 STEMI patients undergoing primary PCI.21 The primary endpoint was 6-month OCT healing score (HS) based on the presence of uncovered or malapposed stent struts and intraluminal filling defects (1.74 (2.39) versus 2.80 (4.44); Pnoninferiority=0.001). This was a small trial powered for non-inferiority of an imaging endpoint. Additionally, only 10 % of STEMI patients during enrollment period were randomized. Therefore, although STEMI lesions are an important target for BVS, we do not currently have enough data to support its use.
Several meta-analyses of randomized trials showed results similar to individual trials, which included higher numerical TLF and stent thrombosis for the Absorb BVS, though most were not statistically significant because of the small numbers of events. A meta-analysis of all the randomized trials, which comprised 3,738 patients, showed similar TLR (OR 0.97; 95 % CI [0.66–1.43]; p=0.87), TLF (OR 1.20; 95 % CI [0.90– 1.60]; p=0.21), and death (OR 0.95; 95 % CI [0.45–2.00]; p=0.89) between BVS and Xience. However, patients who received BVS had higher risk of definite or probable device thrombosis (OR 1.99; 95 % CI [1.00–3.98]; p=0.05).22 Another patient-level meta-analysis of four ABSORB trials (TROFI II was excluded because of the STEMI-only population) found MACE (RR 1.08; p=0.38) and TLF (RR 1.22; p=0.17) to be similar, but a trend for higher device thrombosis (RR 2.09; p=0.08). TVR MI was significantly higher in the BVS group (RR 1.45; p=0.04).23
US Applications
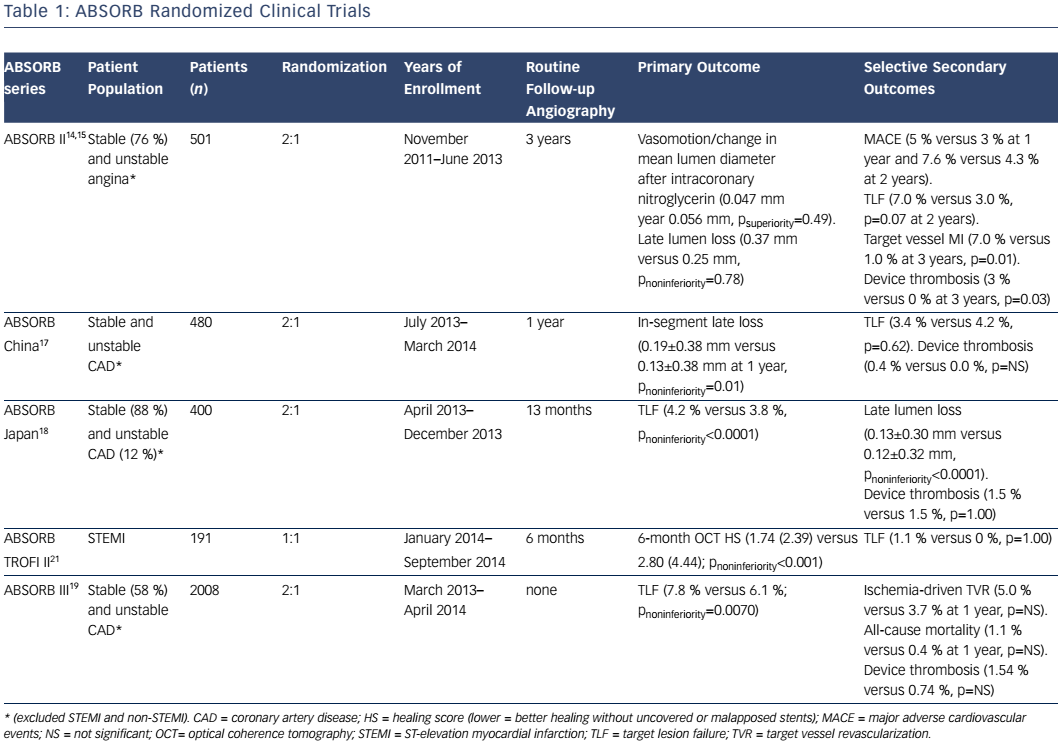
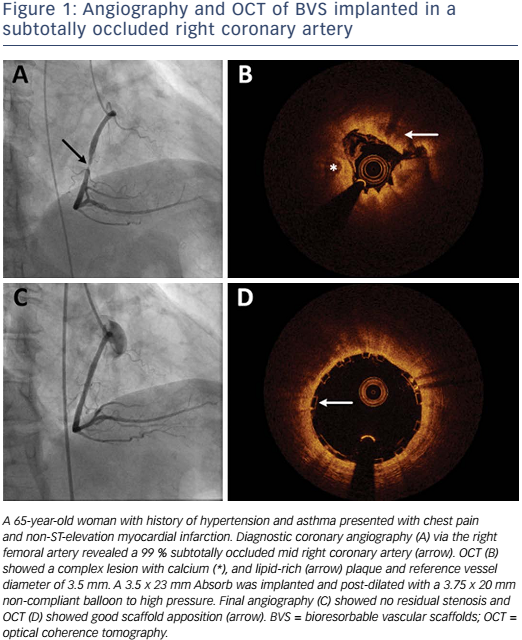
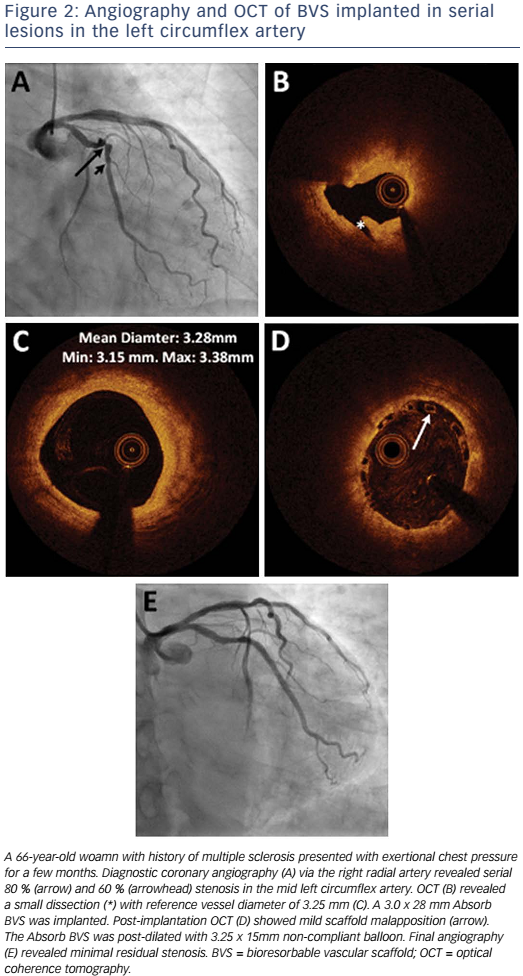
Absorb is the only first-generation BVS approved in the US by the FDA (July 5, 2016) with the following labelling: “The Absorb BVS is a temporary scaffold that will fully resorb over time and is indicated for improving coronary luminal diameter in patients with ischemic heart disease due to de novo native coronary artery lesion (length ≤24 mm) with RVD of ≥2.5 mm and ≤3.75 mm.” Patient and lesion selection are important during the early adoption of this new technology.20 US operators are likely to start with simple lesions and select patients most likely to benefit from a fully resorbable device as Absorb is first introduced into general practice. Figures 1 and 2 are examples of lesions treated in clinical practice in the US. With increasing operator experience over time, BVS may be used to treat more complex lesions while more data are collected. As discussed, in subgroup analysis of ABSORB III, 1-year TLF, its components, and scaffold thrombosis were all higher in the Absorb group compared with the control group in vessels <2.25 mm. Given the potential mismatch between visual estimate and intracoronary imaging, the manufacturer strongly advises using QCA or intracoronary imaging for vessels visually ≤2.75 mm to confirm RVD ≥2.5 mm. The FDA panel also advocates quantitative measurements for vessels ≤3.0 mm to ensure appropriate sizing. Figure 3 shows an example of OCT vessel sizing of a 4.5 mm proximal RCA lesion that was too large for the Absorb BVS. The manufacturer labelling further recommends additional pre-dilation in all lesions with residual stenosis of 20–40 % prior to BVS implantation, and post-dilation with non-compliant balloon at >16 mmHg to ensure adequate scaffold apposition.
There is evidence that these strategies of careful lesion selection and deployment technique can improve outcomes. For example, one mechanism for very late ScT appears to be because of resorption of initially malapposed scaffold struts, causing thrombosis because of mechanical disruption.24,25 In a large registry study of 1,305 consecutive Absorb implants, ScT was 1.8 % at 30 days and 3.0 % at 1 year.26 Lower post-procedure minimum lumen and reference vessel diameters (RVD) were hallmarks of late ScT, and nine of 42 cases occurred after discontinuation of DAPT. Based on early results, a ‘BVS-specific protocol’ was designed to address incomplete BVS expansion with the following steps: 1) predilation with non-compliant balloon to the RVD, 2) BVS implantation only if the predilation balloon is fully expanded, 3) implantation of BVS to RVD, and 4) post-dilation with non-compliant balloon up to a maximum of 0.5 mm larger than RVD. The study showed that after the ‘BVS-specific protocol’ was adopted, ScT rates fell from 3.3 % to 1.0 % at 1 year, suggesting that ScT could be decreased by using careful, regimented implantation techniques. It is reasonable to speculate that such a strategy may have reduced event rates in ABSORB II and ABSORB III and clearly should be an integral part of the procedure for early adoption
An additional unanswered question for BVS implants is the duration of DAPT. All patients in the ABSORB studies were maintained on P2Y12 inhibitor for 12 months and aspirin indefinitely. The 2016 updated DAPT guidelines recommend at least 6 months of DAPT for DES in stable coronary artery disease and at least 12 months for DES in acute coronary syndrome.27 Longer duration is also reasonable based on results from the DAPT trial.28 There are no specific guidelines for DAPT duration after BVS implantation, although with concerns for higher very late ScT rate in clinical trials combined with the duration of scaffold resorption, prolonged DAPT should be strongly considered. It seems reasonable that initial experience in the US should be used in patients with relatively low bleeding risk and eligible for prolonged DAPT.
Lesion Complexity and Characteristics
Lesion complexity is a major consideration for BVS implantation. Exclusion criteria in the ABSORB randomized trials were numerous, including, but not limited to, heavily calcified lesions, extreme angulation or tortuosity, left main or ostial lesions, bifurcation lesions, thrombotic lesions, and chronically occluded lesions. Although there are reports of real-practice experience and registry data on these more complex lesions from outside the US, these were usually reported by operators with substantial device experience and with meticulous technique.29–32 It seems advisable for the initial US experience to avoid more complex lesions such as heavily calcified lesions, bifurcation lesions, and chronic total occlusions. There should be a strategy to advance to higher complexity procedures after demonstrating favorable outcomes with simple lesions and with a commitment for systematic collection of outcomes data in the US.
Conclusion
The BVS is a new category of device that has the potential for improving the treatment of coronary artery disease. The technology offers considerable promise, but as a first-generation device, BVS has been hampered with safety concerns. Additionally, the theoretical longterm benefits have yet to be proven. Adverse outcomes in early trials and practice appear to be related to implantation in smaller vessels or inadequate vessel preparation and lack of post-dilation. It seems reasonable that if the safety concerns can be mitigated by careful lesion sizing and selection in addition to improved implantation technique, the long-term benefit of a fully resorbed stent would become evident.
During initial BVS adoption in the US, physicians should be conservative with careful patient and lesion selection and follow meticulous and regimented implantation techniques involving frequent intracoronary imaging for sizing, and pre- and post-dilation. This should include full adoption of the 4-step BVS specific implantation protocol as described above. Physicians should be cautious not to overextend treatment to complex lesions until there has been substantial experience and demonstration of favorable safety. For now, treatment also should be limited to patients who can comply with at least 12 months of DAPT and longer therapy should be considered until the mechanisms of very late ScT are more completely understood. While we await the next generation of BVS, which likely will have thinner struts, more consistent deployment results and faster resorption, we can thoughtfully proceed with using the current device as approved and recommended. The goal of ‘nothing left behind’ in a repaired and normally functioning coronary artery remains an important one.